getting jumpy are you?
oh btw, looks like one of your packs is out of order. Originally Posted by dilbert firestorm
that's what he gets for cloning his pac's. replication failure.
BAHHAHAAA
The proximal origin of SARS-CoV-2
The proximal origin of SARS-CoV-2
- Correspondence
- Published: 17 March 2020
Nature Medicine volume 26, pages450–452(2020)Cite this article
- Kristian G. Andersen,
- Andrew Rambaut,
- W. Ian Lipkin,
- Edward C. Holmes &
- Robert F. Garry
- 4.95m Accesses
- 475 Citations
- 34732 Altmetric
- Metrics details
To the Editor — Since the first reports of novel pneumonia (COVID-19) in Wuhan, Hubei province, China1,2, there has been considerable discussion on the origin of the causative virus, SARS-CoV-23 (also referred to as HCoV-19)4. Infections with SARS-CoV-2 are now widespread, and as of 11 March 2020, 121,564 cases have been confirmed in more than 110 countries, with 4,373 deaths5.
SARS-CoV-2 is the seventh coronavirus known to infect humans; SARS-CoV, MERS-CoV and SARS-CoV-2 can cause severe disease, whereas HKU1, NL63, OC43 and 229E are associated with mild symptoms6. Here we review what can be deduced about the origin of SARS-CoV-2 from comparative analysis of genomic data. We offer a perspective on the notable features of the SARS-CoV-2 genome and discuss scenarios by which they could have arisen. Our analyses clearly show that SARS-CoV-2 is not a laboratory construct or a purposefully manipulated virus.
Notable features of the SARS-CoV-2 genome
Our comparison of alpha- and betacoronaviruses identifies two notable genomic features of SARS-CoV-2: (i) on the basis of structural studies7,8,9 and biochemical experiments1,9,10, SARS-CoV-2 appears to be optimized for binding to the human receptor ACE2; and (ii) the spike protein of SARS-CoV-2 has a functional polybasic (furin) cleavage site at the S1–S2 boundary through the insertion of 12 nucleotides8, which additionally led to the predicted acquisition of three O-linked glycans around the site.
1. Mutations in the receptor-binding domain of SARS-CoV-2
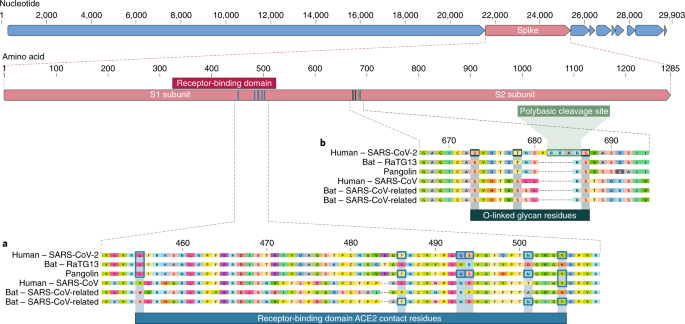
The receptor-binding domain (RBD) in the spike protein is the most variable part of the coronavirus genome1,2. Six RBD amino acids have been shown to be critical for binding to ACE2 receptors and for determining the host range of SARS-CoV-like viruses7. With coordinates based on SARS-CoV, they are Y442, L472, N479, D480, T487 and Y4911, which correspond to L455, F486, Q493, S494, N501 and Y505 in SARS-CoV-27. Five of these six residues differ between SARS-CoV-2 and SARS-CoV (Fig. 1a). On the basis of structural studies7,8,9 and biochemical experiments1,9,10, SARS-CoV-2 seems to have an RBD that binds with high affinity to ACE2 from humans, ferrets, cats and other species with high receptor homology7.
Fig. 1: Features of the spike protein in human SARS-CoV-2 and related coronaviruses.
a, Mutations in contact residues of the SARS-CoV-2 spike protein. The spike protein of SARS-CoV-2 (red bar at top) was aligned against the most closely related SARS-CoV-like coronaviruses and SARS-CoV itself. Key residues in the spike protein that make contact to the ACE2 receptor are marked with blue boxes in both SARS-CoV-2 and related viruses, including SARS-CoV (Urbani strain). b, Acquisition of polybasic cleavage site and O-linked glycans. Both the polybasic cleavage site and the three adjacent predicted O-linked glycans are unique to SARS-CoV-2 and were not previously seen in lineage B betacoronaviruses. Sequences shown are from NCBI GenBank, accession codes MN908947, MN996532, AY278741, KY417146 and MK211376. The pangolin coronavirus sequences are a consensus generated from SRR10168377 and SRR10168378 (NCBI BioProject PRJNA573298)29,30.
Full size image
While the analyses above suggest that SARS-CoV-2 may bind human ACE2 with high affinity, computational analyses predict that the interaction is not ideal7 and that the RBD sequence is different from those shown in SARS-CoV to be optimal for receptor binding7,11. Thus, the high-affinity binding of the SARS-CoV-2 spike protein to human ACE2 is most likely the result of natural selection on a human or human-like ACE2 that permits another optimal binding solution to arise. This is strong evidence that SARS-CoV-2 is not the product of purposeful manipulation. Originally Posted by matchingmole
The proximal origin of SARS-CoV-2Didn't explain much how it entered the Human Population.
The proximal origin of SARS-CoV-2
- Correspondence
- Published: 17 March 2020
Nature Medicine volume 26, pages450–452(2020)Cite this article
- Kristian G. Andersen,
- Andrew Rambaut,
- W. Ian Lipkin,
- Edward C. Holmes &
- Robert F. Garry
- 4.95m Accesses
- 475 Citations
- 34732 Altmetric
- Metrics details
To the Editor — Since the first reports of novel pneumonia (COVID-19) in Wuhan, Hubei province, China1,2, there has been considerable discussion on the origin of the causative virus, SARS-CoV-23 (also referred to as HCoV-19)4. Infections with SARS-CoV-2 are now widespread, and as of 11 March 2020, 121,564 cases have been confirmed in more than 110 countries, with 4,373 deaths5.
SARS-CoV-2 is the seventh coronavirus known to infect humans; SARS-CoV, MERS-CoV and SARS-CoV-2 can cause severe disease, whereas HKU1, NL63, OC43 and 229E are associated with mild symptoms6. Here we review what can be deduced about the origin of SARS-CoV-2 from comparative analysis of genomic data. We offer a perspective on the notable features of the SARS-CoV-2 genome and discuss scenarios by which they could have arisen. Our analyses clearly show that SARS-CoV-2 is not a laboratory construct or a purposefully manipulated virus.
Notable features of the SARS-CoV-2 genome
Our comparison of alpha- and betacoronaviruses identifies two notable genomic features of SARS-CoV-2: (i) on the basis of structural studies7,8,9 and biochemical experiments1,9,10, SARS-CoV-2 appears to be optimized for binding to the human receptor ACE2; and (ii) the spike protein of SARS-CoV-2 has a functional polybasic (furin) cleavage site at the S1–S2 boundary through the insertion of 12 nucleotides8, which additionally led to the predicted acquisition of three O-linked glycans around the site.
1. Mutations in the receptor-binding domain of SARS-CoV-2
The receptor-binding domain (RBD) in the spike protein is the most variable part of the coronavirus genome1,2. Six RBD amino acids have been shown to be critical for binding to ACE2 receptors and for determining the host range of SARS-CoV-like viruses7. With coordinates based on SARS-CoV, they are Y442, L472, N479, D480, T487 and Y4911, which correspond to L455, F486, Q493, S494, N501 and Y505 in SARS-CoV-27. Five of these six residues differ between SARS-CoV-2 and SARS-CoV (Fig. 1a). On the basis of structural studies7,8,9 and biochemical experiments1,9,10, SARS-CoV-2 seems to have an RBD that binds with high affinity to ACE2 from humans, ferrets, cats and other species with high receptor homology7.
Fig. 1: Features of the spike protein in human SARS-CoV-2 and related coronaviruses.
a, Mutations in contact residues of the SARS-CoV-2 spike protein. The spike protein of SARS-CoV-2 (red bar at top) was aligned against the most closely related SARS-CoV-like coronaviruses and SARS-CoV itself. Key residues in the spike protein that make contact to the ACE2 receptor are marked with blue boxes in both SARS-CoV-2 and related viruses, including SARS-CoV (Urbani strain). b, Acquisition of polybasic cleavage site and O-linked glycans. Both the polybasic cleavage site and the three adjacent predicted O-linked glycans are unique to SARS-CoV-2 and were not previously seen in lineage B betacoronaviruses. Sequences shown are from NCBI GenBank, accession codes MN908947, MN996532, AY278741, KY417146 and MK211376. The pangolin coronavirus sequences are a consensus generated from SRR10168377 and SRR10168378 (NCBI BioProject PRJNA573298)29,30.
Full size image
While the analyses above suggest that SARS-CoV-2 may bind human ACE2 with high affinity, computational analyses predict that the interaction is not ideal7 and that the RBD sequence is different from those shown in SARS-CoV to be optimal for receptor binding7,11. Thus, the high-affinity binding of the SARS-CoV-2 spike protein to human ACE2 is most likely the result of natural selection on a human or human-like ACE2 that permits another optimal binding solution to arise. This is strong evidence that SARS-CoV-2 is not the product of purposeful manipulation. Originally Posted by matchingmole
 Originally Posted by HoeHummer
Originally Posted by HoeHummer
